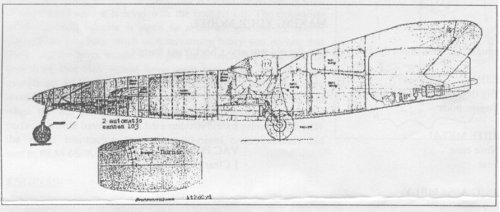
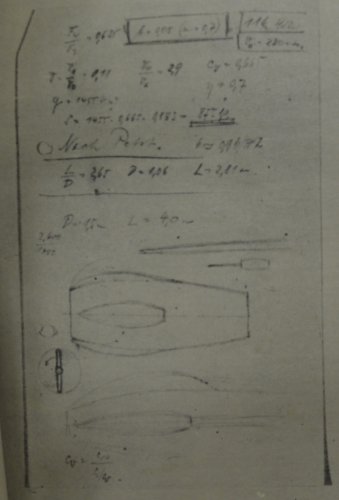
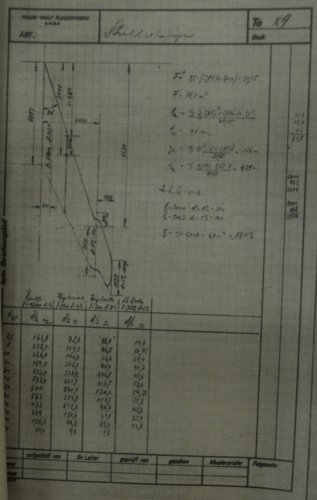
The handwriting under the printing "Bez." (short for "Bezeichnung" = designation) may indeed
say "Strahlschnellbomber" (Fast jet bomber), but it's quite difficult to read. "F" may stand for
wing area (134 m²), given, too is "F1" as 10 m², and F/F1 as 13.4. Mathematically correct, but in
the moment, I have no idea, what F1 actually is. Maybe the area of the tailplane ? "GST" could be
"Startgewicht" /take-off weight (25,000 kg) and another weight data (G) is given as 20,000 kg,
obviously used for calculating wing loading, which is quite correctly given as 150 kg/m².
Fuel could have been 8,000 kg and bomb load 3,000 kg.
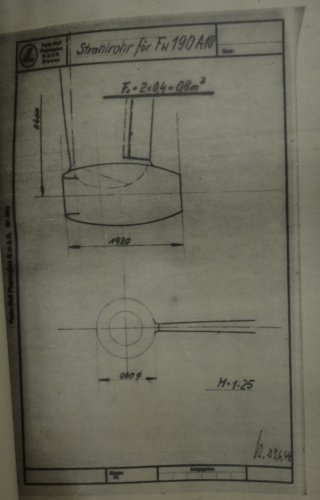
The sketch is drawn on a "Festigkeitsberechnungblatt" (sheet for stress analyses. It obviously
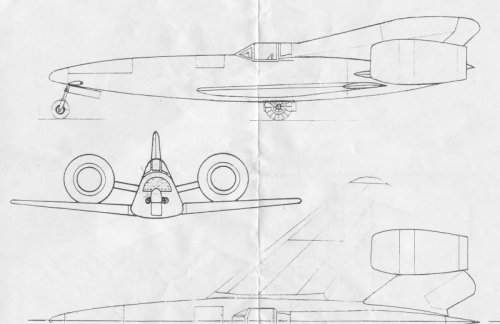
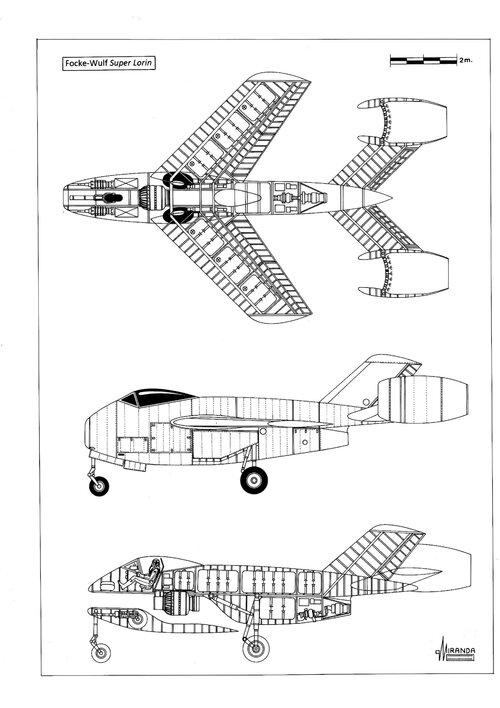
isn't that, but to my opinion a just very rough calculation of an enlarged derivative of the Ta 283.